Performance evaluation, post-market surveillance and the IVDR

Performance evaluation, in accordance with Article 56 and Annex XIII, including the new post-market performance follow-up (PMPF) plan must be defined within the QMS. This is an area that will have variation based on the classification of the device and should be adapted accordingly. The QMS process therefore needs to be sufficiently descriptive while allowing the necessary variation to accommodate a manufacturer’s different types of devices. The PMPF will be a key element in the post-market surveillance activities. The plan may be a stand-alone document, or if it is sufficiently homogenous across a product family, can be part of the QMS as either a procedure or a template to be completed for each product. If it is intended to be a procedure, the scope of the procedure will need to be sufficiently clear to identify it as the plan. The introduction of the plan is one of the strong indications that the IVDR is shifting from reactive PMS (relying only on vigilance activities) to something more proactive.
Post-market surveillance activities must be established, implemented and maintained in accordance with Article 78, and, as stated in the IVDR, this must be an integral part of the QMS (see Figure 1). It is necessary to note that a number of links between post-market surveillance activities and other areas of the QMS have been prescribed in the regulation, including risk management, performance evaluation, updates to the technical documentation, Corrective and Preventive Actions (CAPA), adverse event reporting and process/product monitoring. For IVDs in class C and D, the manufacturer shall prepare a Periodic Safety Update Report (PSUR). Ultimately, the manufacturer needs to demonstrate that the product is compliant with all safety and performance requirements, using as reference any Common Specifications (CS) that exist or ‘state-of-the-art’ comparisons.
Figure 1 – Post-market surveillance throughout the product life-cycle.
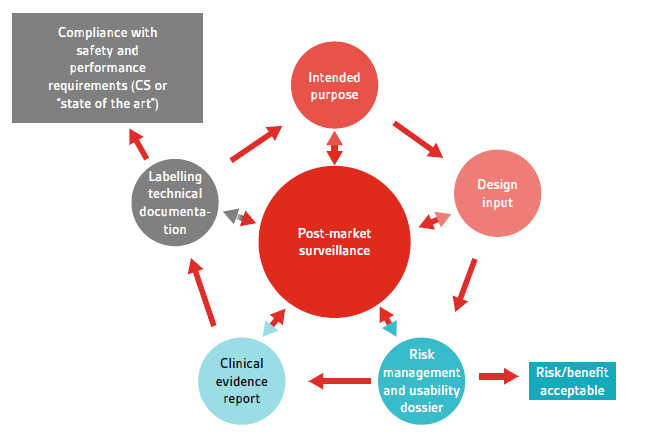
The manufacturer needs to take into account these links (as inputs, outputs or connections) to the various processes in order to ensure that they are adequately interfaced and effectively associated. A key aspect of this will be in ensuring that common terminology and references are used throughout the various files in order to maintain traceability. A common source of error can be a disconnect between early product development, before the name of the final product has been established, and the final marketed name of the product. Manufacturers must ensure that these are adequately connected, if not fully updated, in order to maintain the connection throughout the life-cycle of the device.
This is an excerpt from the white paper Developing and maintaining a quality management system for IVDs. To download our other medical device white papers, please visit the Insight page on the Compliance Navigator website.
Request more information today for a call back from a member of our sales team so that you can get a better understanding of how Compliance Navigator can meet your needs.
The Compliance Navigator blog is issued for information only. It does not constitute an official or agreed position of BSI Standards Ltd or of the BSI Notified Body. The views expressed are entirely those of the authors.
