IVDR: Practical Considerations for the Performance Evaluation Plan and Report

The IVDR (EU 2017/746) brings new requirements for manufacturers with regard to Performance Evaluation and Clinical Performance Studies and one of those is the need for a Performance Evaluation Plan (PEP) and Performance Evaluation Report (PER).
What is a PEP?
The PEP should be constructed to enable the evaluator to correctly perform and appropriately report on the Performance Evaluation. The PEP should cover at least the following (per the IVDR) or should include a justification for its exclusion:
- details of the intended purpose, or intended use, of the IVD;
- the performance specifications for the IVD, as established by the manufacturer to ensure that the intended purpose is fulfilled;
- the identity of the analyte or marker to be determined by the IVD;
- any related certified reference standards or measurement procedures to allow for metrological traceability;
- the clearly identified specific target patient groups for the IVD along with indications, limitations and contra-indications;
- the relevant GSPRs that the Performance Evaluation will support;
- details of the methodology, including appropriate statistical tools, to be used in the assessment of the analytical and clinical performance of the IVD and the limitations of the device and information provided by it;
- a description of the state of the art relating to the IVD and including any relevant standards, common specifications, guidance documents, etc.;
- the parameters to be used (based on the state of the art in medicine) to determine the acceptability of the benefit-risk ratio for the intended purpose/s and for the analytical and clinical performance of the IVD;
- an overview of each of the development phases for the IVD with milestones, acceptance criteria and timing of verification and validation testing;
- if the IVD is software used as a medical device, then details of reference databases and other sources of data used as the basis for the device’s decision making;
- the process for PMPF planning.
Taking time to prepare the PEP with sufficient detail – as with any experimental protocol or research plan – will improve both efficiency and effectiveness of the Performance Evaluation process when it is conducted. It is likely that a number of stakeholders may need to be engaged to develop, and then implement, the PEP. For smaller companies, some of these may not be immediately available within the organisation and careful consideration of appropriate and timely resourcing should also be part of this Performance Evaluation planning process.
What is a PER?
The PER has its roots in the Clinical Evaluation Report (CER) required under the Medical Device Directive (MDD) and now the Medical Device Regulation (MDR). The PER should be an output from the process of Performance Evaluation as noted in previous blog posts and be populated from the results of applying the Performance Evaluation Plan.
Annex VIII, Part A (1.3.2) of the IVDR outlines the specific components of the PER and specify that it must include:
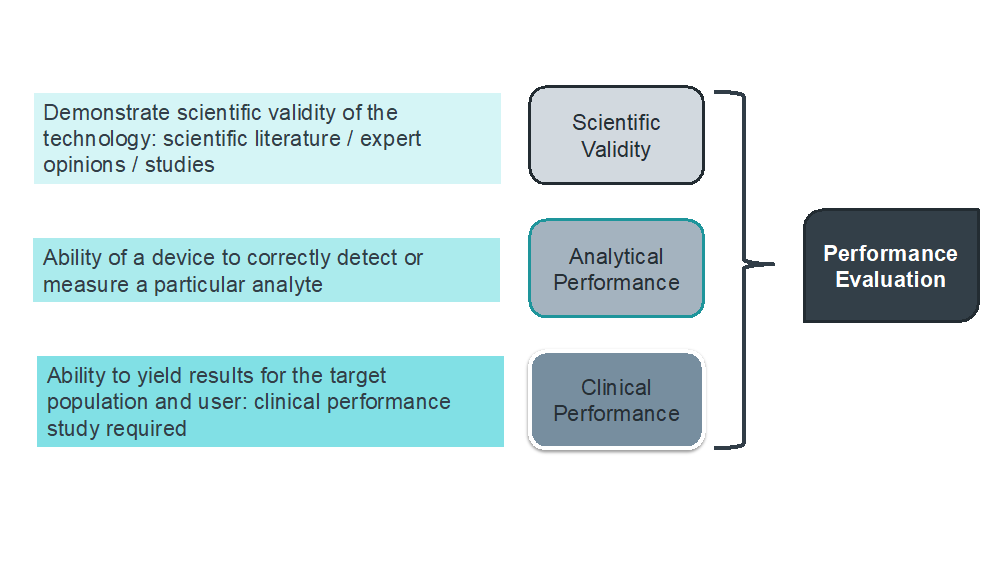
- the scientific validity report;
- the analytical performance report;
- the clinical performance report; and
- an assessment of those reports supporting the demonstration of the clinical evidence as sufficient to make a decision on the benefit-risk ratio.
Other aspects that the PER should cover are the reasoning behind the clinical evidence gathering methods used including any literature searches (and their related protocols and reports); a description of the technology behind the IVD; the intended purpose and associated claims; the actual scientific validity and the analytical and clinical performance data that has been evaluated; the clinical evidence supporting the use of the device when assessed in the context of the current state of the art in medicine; and any new conclusions coming from PMPF or other sources.
This means that the PER is likely to contain all of the device related analytics such as sensitivity, specificity, reproducibility, stability, and so on. Those companies that have prior experience with FDA 510(k) submissions for their devices may already have some elements o fthe PER in place. However, as the PER must support the clinical benefit of the IVD test, there will inevitably be data that will needed to be generated and gathered as appropriate.
Performance Evaluation Reports for Class C and D devices must be updated at least annually, whereas PERs for Class A and B devices should be updated as needed, although at least a three years review cycle would be recommended.
Practical Considerations
From the above it can be seen that there are a number of key aspects that should be considered in the Performance Evaluation process, but in particular the three areas of scientific validity, analytical performance and clinical performance.
In each of these areas, whether the devices in question are already on the market or those in development, the manufacturer should begin by developing a gap analysis that will answer some basic questions, such as:
What technology is the device based on and how does it compare to the current state of the art of scientific knowledge?
- How much research has been conducted on the device and its intended purpose?
- Is this research by the manufacturer, by an academic institution, by an independent evaluator, etc.?
- Have proof of concept studies been conducted to the right level of good laboratory practice and are the results sufficiently robust?
- What analytical performance testing has been carried out?
- What standards or references have been used for these tests?
- Do the results provide a high level of assurance?
Based on the answers to these and other questions specific to the IVD and intended performance in question, the manufacturer will be able to develop a detailed picture that will inform the Performance Evaluation Plan. Alongside this technical gap analysis and action plan the manufacturer should also develop a detailed financial feasibility analysis to ensure that the correct levels of resources are available at the appropriate time for the Performance Evaluation and associated activities such as clinical data gathering by means of a Clinical Performance Study. The latter will be discussed in more detail in a future blog post.
For manufacturers with IVDs already on the market or those with device in development, time is short. The Date of Application (May 2022) is a hard deadline for these devices to have achieved their certification under the IVDR. Therefore, sufficient time needs to be factored in for on-site QMS audit to the IVDR, and reviews of required technical documentation. Bearing in mind that there are a significant volume of these devices on the market, and the capacity of NBs will not be infinite, it would seem prudent to allow at least 12 months from application to certification. Therefore, it is vital that a manufacturer starts this process as soon as possible to ensure all appropriate additional testing and data may be gathered in sufficient time. So don’t wait – if you haven’t already started this process, start now!
Author: Jacques du Preez, COO and Robin Stephens, CEO, Psephos Biomedica
The Compliance Navigator blog is issued for information only. It does not constitute an official or agreed position of BSI Standards Ltd or of the BSI Notified Body. The views expressed are entirely those of the authors.
