Technical Documentation
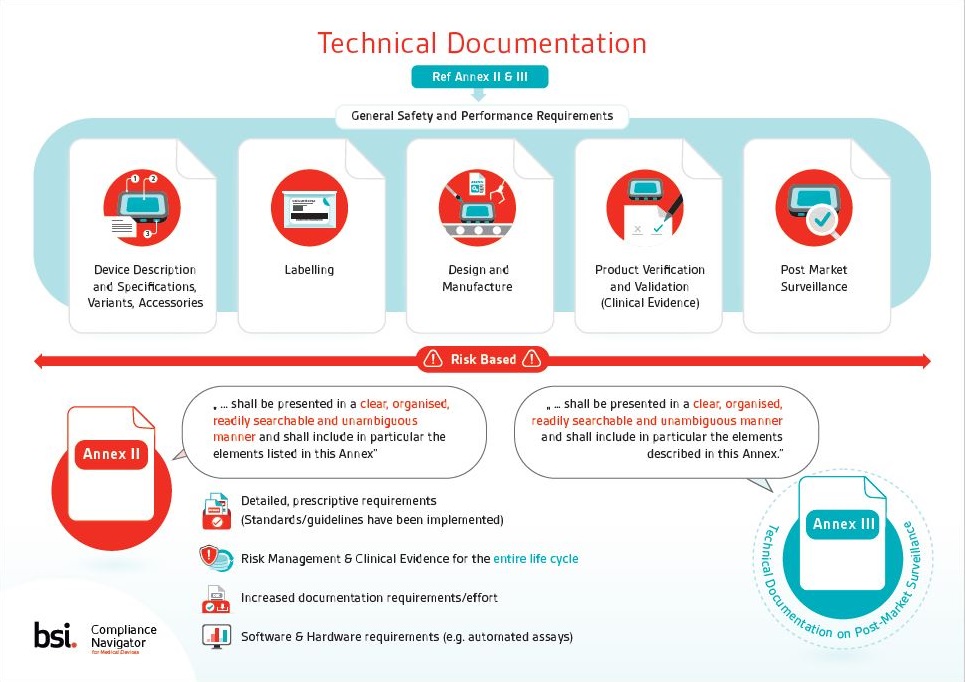
Download this free IVDR technical documentation infographic for insight into annexes II and III, general safety and performance requirements (GSPRs) and harmonized standards related to GSPRs.
Understand regulatory and technological changes with our white papers, infographics and more.
Download this free IVDR technical documentation infographic for insight into annexes II and III, general safety and performance requirements (GSPRs) and harmonized standards related to GSPRs.

The International Medical Device Regulators Forum (IMDRF) aims to accelerate international medical device regulatory convergence.Through the IMDRF, regulators reached consensus on what software is considered a medical device. Regulators call it ‘software as a medical device’ (SaMD). This paper provides a comparison of how SaMD is regulated in the US and in the EU.

The conduct of a clinical investigation is one of the most time consuming and resource intensive activities that a medical device manufacturer can face. This paper discusses important new requirements for pre-market and post-market clinical investigations under the European MDR.
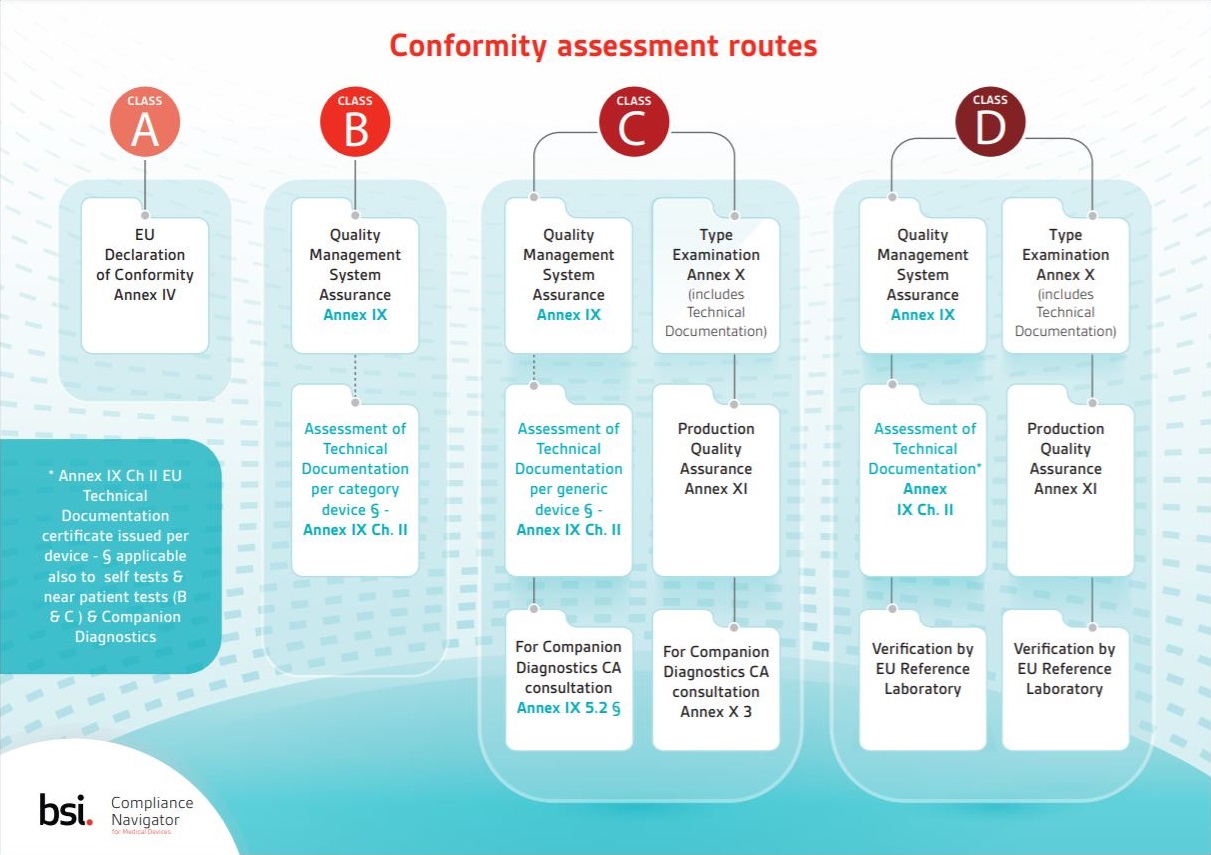
Learn about the different IVDR conformity assessment routes available for IVD devices falling into each risk class with this Compliance Navigator infographic.
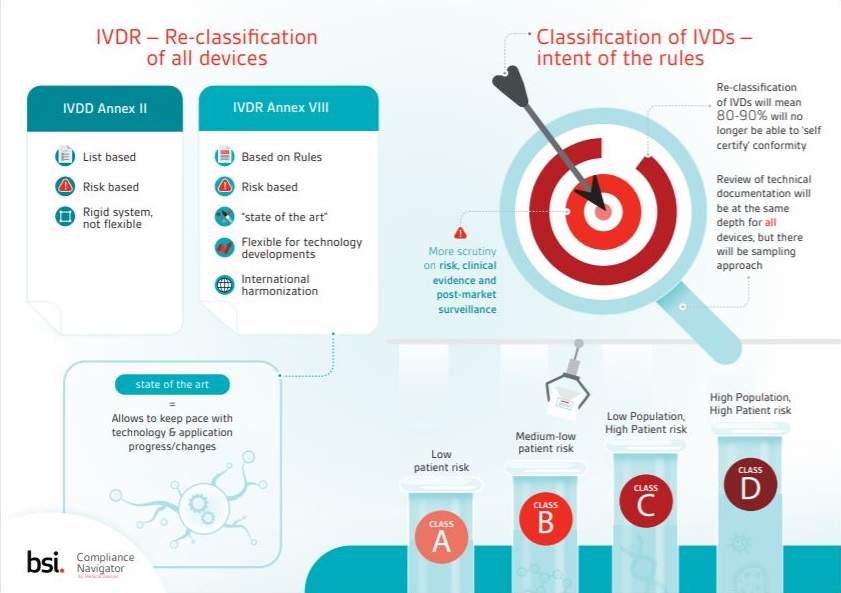
Download this infographic about classification and the IVDR and understand key changes to IVD classification brought about by the Regulation, learn about the classification rules and view lists of examples for each risk class.
The US FDA, Health Canada and BSI have published a number of freely available resources related to the COVID-19 pandemic.

How is AI different from traditional medical devices and medical software and what are the implications of those differences? What controls are necessary to ensure AI in healthcare is safe and effective?
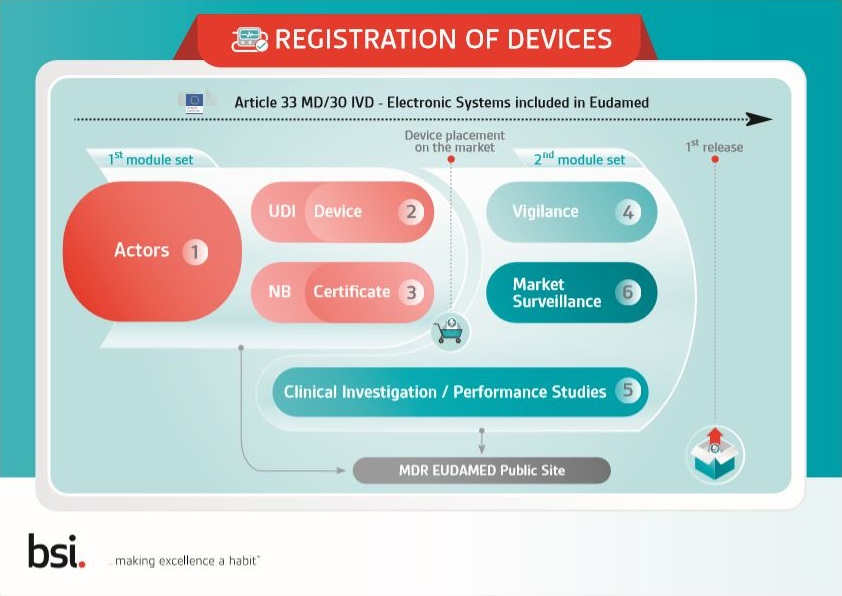
Learn about unique device identification (UDI) and EUDAMED with this free infographic.

Regulatory requirements for medical devices include particular requirements for devices supplied or intended to be used in a sterile state. These regulatory requirements have been supported by a portfolio of standards. This paper provides an overview of these regulatory requirements and the standards that support them.

Find out how post market surveillance requirements are changing from the MDD/AIMDD to the MDR.

Gain insight into the concept of 'significant change' under Article 120(3) of the Regulation.
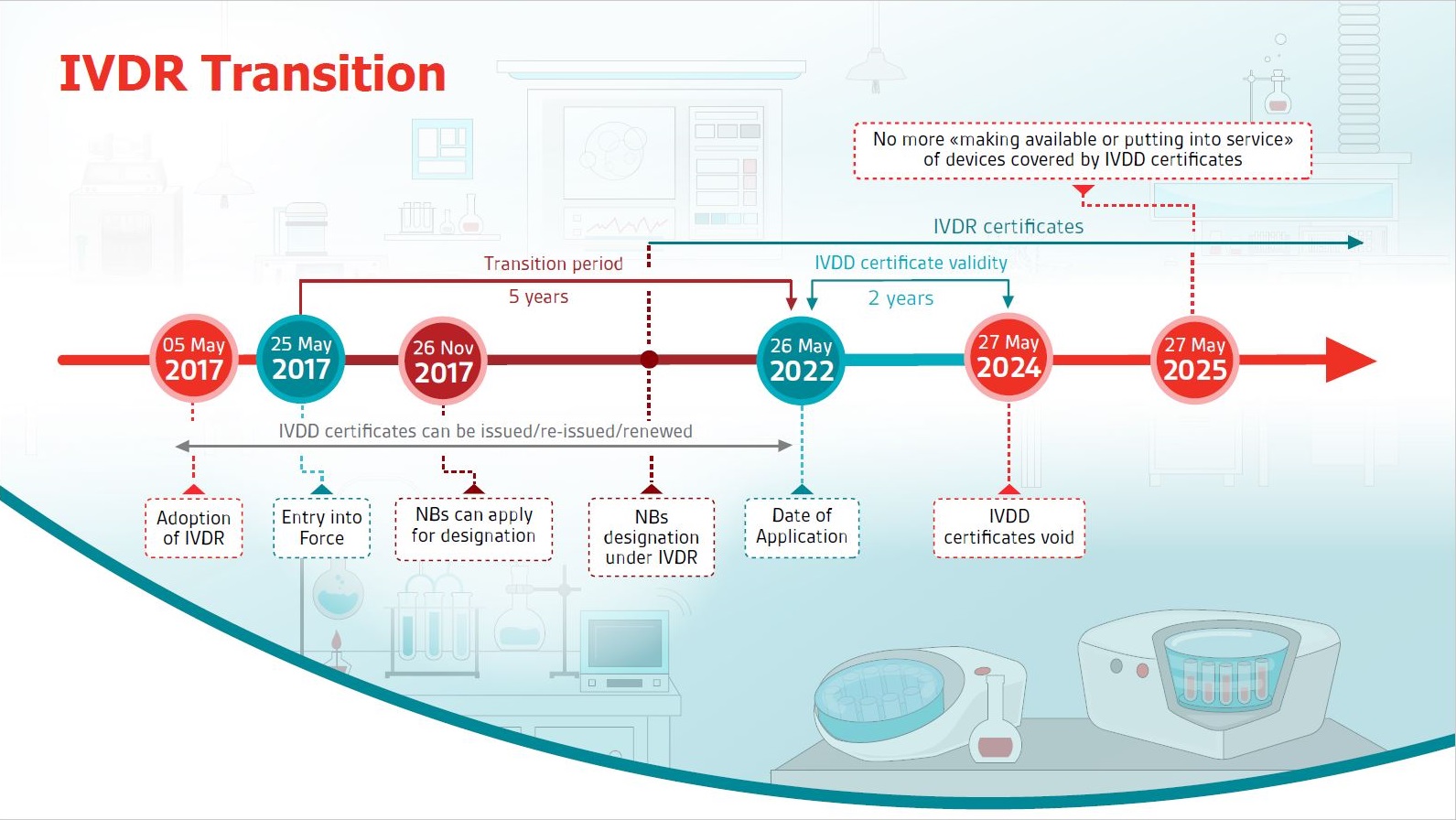
Learn about key dates of the IVDR transition with this free Compliance Navigator infographic.

Re-classification of IVDs will mean 80-90% will no longer be able to ‘self certify’ conformity.
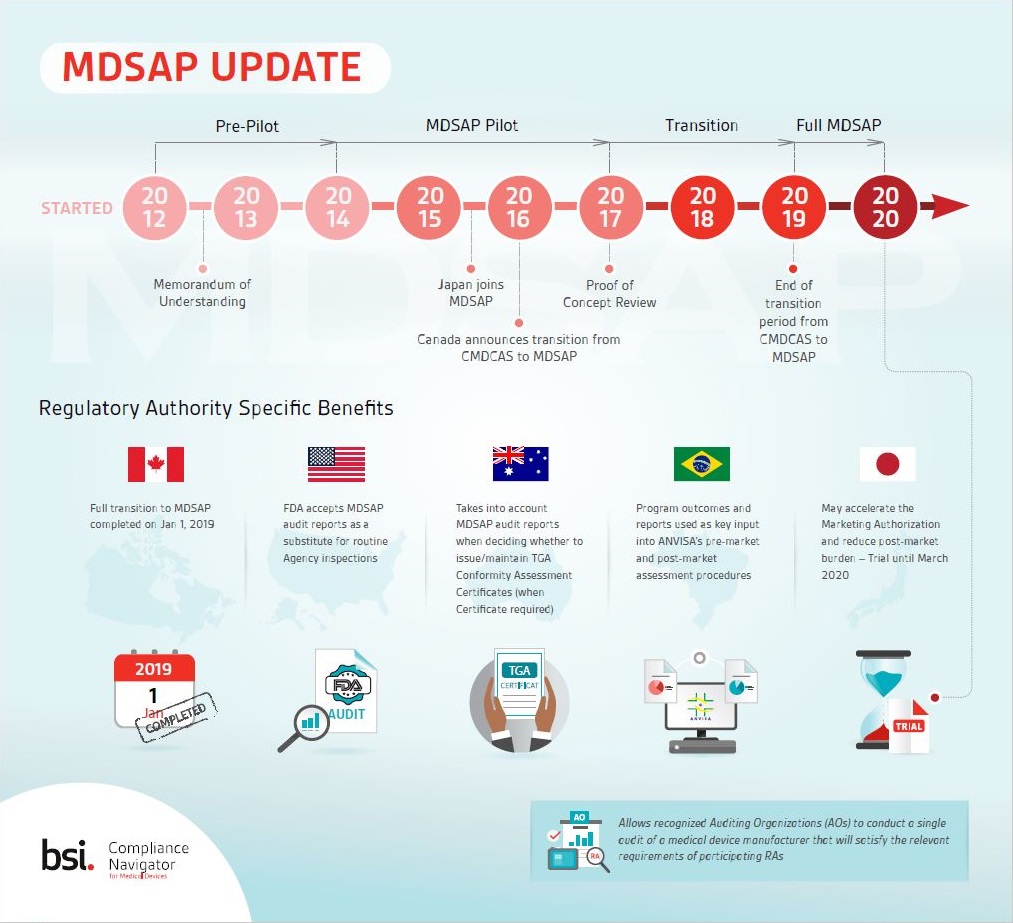
Remind yourself of some key MDSAP milestones as well as several of the benefits to manufacturers that are specific to different participating regulatory authorities.
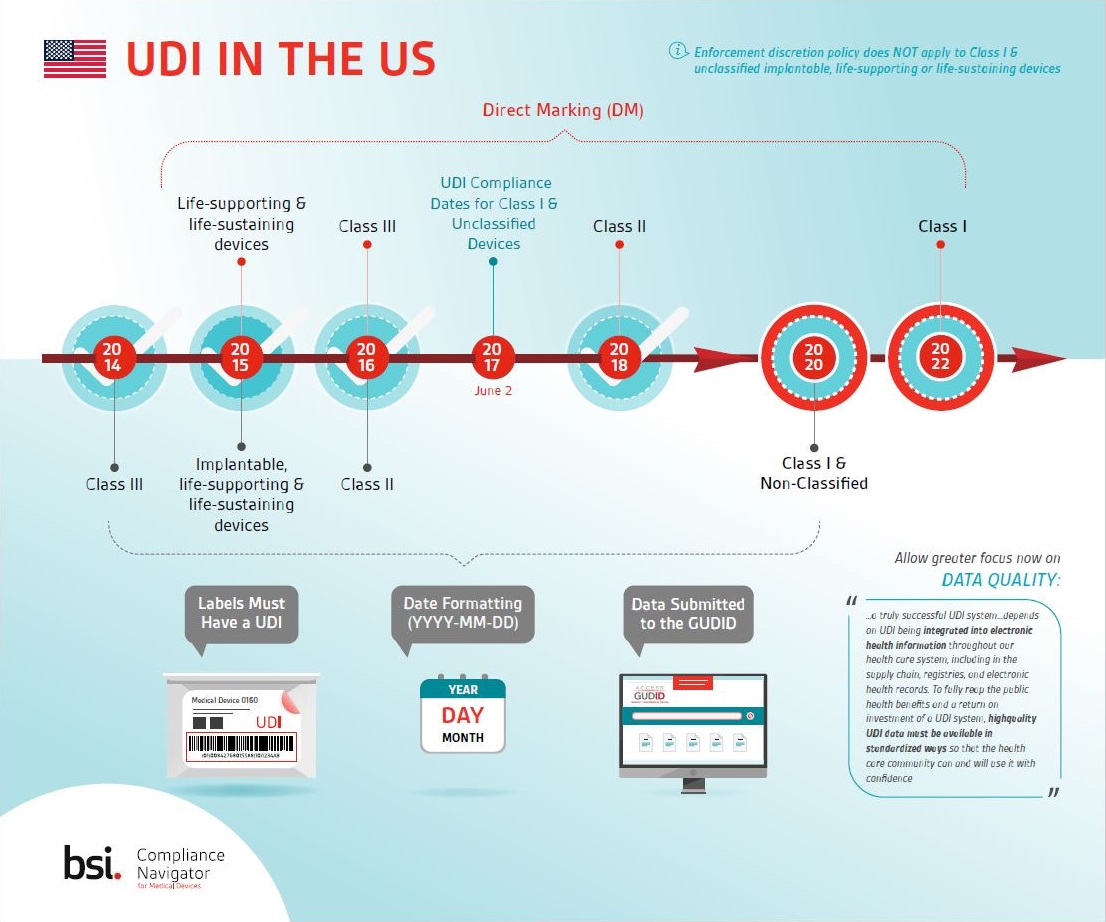
Learn about key milestones for UDI in the US and download this Compliance Navigator infographic.
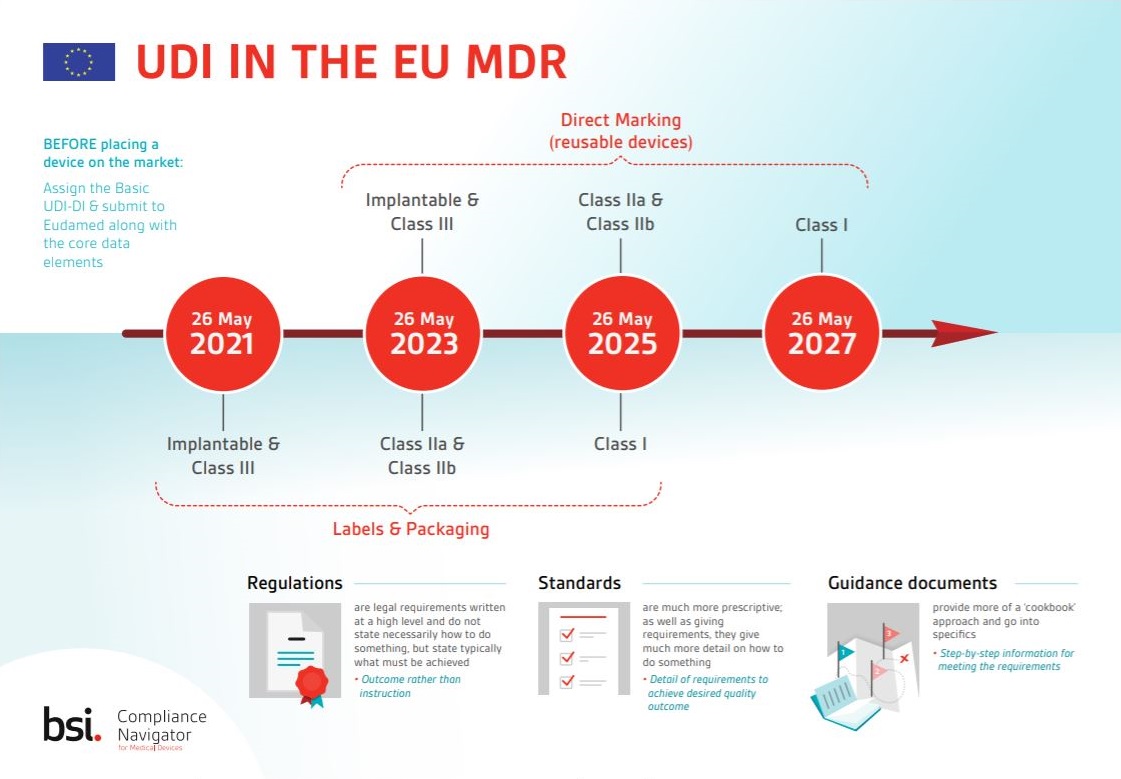
Learn about important dates for labels and packaging requirements and get an overview of the key requirements of Article 27 of the EU MDR.

This infographic on the new ISO 14971 gives an overview of new and adapted definitions of key terms, the use and misuse of medical devices and the risk management process as set out in the third edition of this medical devices risk management standard.

Download this BSI medical devices white paper, authored by Mika Reinikainen and Dr Maurizio Suppo, for a historical overview of the development of medical device and IVD device classification; an explanation of the new IVDR classification rules; and an analysis of the implications of these new rules.

Gain insight into post-market surveillance for medical devices and in vitro diagnostic devices under the EU Regulations with this free download.

Download this BSI medical devices white paper, authored by Prof. Kenny Dalgarno, for a review of the history of 3D printing of medical devices, a discussion of the key characteristics of this technology's successful exploitation and an examination of the scope for bioprinting processes to enhance medical devices, bearing in mind the lessons learnt from the more established 3D printing industry

Download this free white paper for a discussion of the development of risk management over the past centuries; the risk management process as described in ISO 14971, including a discussion of the main changes in the third edition, which is expected to be published in 2019; and the broader context of ISO 14971 and its use in conjunction with other international standards to demonstrate compliance with regulatory requirements.

Medical Device Single Audit Program (MDSAP) allows a single audit of a medical device manufacturers QMS which satisfies the requirements of multiple regulatory jurisdictions. Audits are conducted by Auditing Organizations (AO), such as BSI, authorized by the participating Regulatory Authorities (RA) to audit under MDSAP requirements.

This white paper provides a very brief overview of where AI is being used in healthcare, and why it might be increasingly seen in medical devices. We also consider what specific additional requirements this might place on regulatory requirements in the near future.

Download the first installment of our 'MDR and IVDR FAQs Series' for insightful answers to key questions about responsible persons, authorized representatives and the new Regulations.

Download this free guide for a comparison of the annexes of the MDD and the MDR, covering product requirements and declarations of conformity. The guide is an excerpt from the Smart Support series: a series of topic-specific expert commentaries on the MDR/IVDR.

This guide presents a summary of the provisions of some of the articles of the MDD and MDR together with commentary providing discussion and highlighting the key differences. The guide is an excerpt from the Smart Support series: a series of topic-specific expert commentaries on the MDR/IVDR.

Digital technology in the medical devices sector has the power to excite. But, it is important that a mature, critical approach is taken when evaluating new technologies. Download this BSI medical devices white paper today and learn about digital maturity.

Gain expert insight into the background to the new EU rules regarding post-market surveillance (PMS) and the actions your organization may need to take as a result of them. This guide is an excerpt from the Smart Support series: a series of topic-specific expert commentaries on the MDR/IVDR.

Learn about the background to the new EU rules regarding authorized representatives and the actions your organization may need to take as a result of them with our dedicated guide. This guide is an excerpt from the Smart Support series: a series of topic-specific expert commentaries on the MDR/IVDR.

Download our guide and gain expert insight into the background to the new EU rules relating to UDI and the actions your organization may need to take as a result of them. This guide is an excerpt from the Smart Support series: a series of topic-specific expert commentaries on the MDR/IVDR.

Download our guide and gain expert insight into the background to the rules regarding importers and distributors under the MDR/IVDR and the actions your organization may need to take as a result. This guide is an excerpt from the Smart Support series: a series of topic-specific expert commentaries on the MDR/IVDR.

Download our guide to the risk classification rules under the MDR and gain expert insight into the background to the new EU rules regarding risk classification and the actions your organization may need to take as a result of them. This guide is an excerpt from the Smart Support series: a series of topic-specific expert commentaries on the MDR/IVDR.

Download this white paper and learn about the QMS requirements under the new In Vitro Diagnostic Device Regulation (IVDR). The white paper includes chapters on: risk management; performance evaluation and post-market surveillance; unique device identification (UDI); and continuous improvement activities.

The MDR and IVDR represent significant changes to European legislation for medical devices and in vitro diagnostic medical devices (IVDs). One significant new requirement is that manufacturers and authorized representatives for both medical devices and IVDs appoint at least one person responsible for regulatory compliance with responsibilities that cover the quality management system (QMS), regulatory documentation, post-market surveillance and vigilance reporting, and devices used for clinical investigation. Download our guide to learn more about the background to the EU rules regarding responsible persons and the actions your organization may need to take as a result of them.

Nanotechnology is now a mature subject in terms of the basic science, and while there were many claims for what it could achieve 20 years ago, the application to medicine and healthcare has now become more well defined. When considering nanotechnology’s future in the medical devices field, it is best to divide the field into five main categories: medical image enhancement; drug delivery vehicles; nanomaterials for functional coatings; therapeutic aspects of nanomaterials that make use of their small size and properties; and finally biosensors that make use of nanoparticles. There are common themes that apply to each of these fields and in this review we will summarize these and identify gaps in understanding and opportunities.

Before placing a medical device on the European market, manufacturers need to produce technical documentation providing evidence of conformity with the relevant legislation. Technical documentation needs to be in compliance with the Medical Device Regulation. Download this whitepaper to read more.

It's been a year since the final text of the new European Medical Device and In Vitro Diagnostics Regulations was published. The timeline for implementation of the new regulations is shortening with each day. Understand the new MDR and IVDR with our infographics and ensure smooth transition for your company.

This paper outlines the requirements specific to incident reporting, vigilance, mandatory problem reporting, medical device reports and adverse event reporting, herein termed ‘vigilance’, in comparison with the requirements of the recently published European Medical Device Regulation (MDR) to support those working with these aspects within the MDSAP Programme. Manufacturers who wish to supply their devices outside of these regions may have many more requirements to meet, the discussion of which is beyond the scope of this paper.

The purpose of this white paper is to compare the ERs in the MDD and AIMDD to the SPRs in Annex I of the new MDR. Where there are 13 ERs in the MDD and 16 in the AIMDD, there are 23 SPRs in the new MDR. The overall text and requirements are expanded, but the scope and topics are consistent overall with the previous directives with a few notable exceptions.

This paper addresses a number of areas, including PMS as an element of the management of clinical evidence throughout the device lifecycle; the PMS system, which is the comprehensive process used to collect, analyze and take action on PMS information; the PMS plan, which describes the application of the PMS system to a device or device family;preparation of a summary report of PMS information; complaint handling and reporting of vigilance; and, electronic submission of vigilance data and summary reports of PMS.

This paper considers the cybersecurity challenges facing the healthcare sector arising from the convergence of technology, hyper-connectivity and recent developments in regulation. It explains the issues and tensions between safety and security and what can be done to resolve them. The paper highlights emerging good practice and approaches that manufacturers can take to improve medical device security throughout its lifecycle. The paper will also be of interest to others in the sector, including healthcare providers, IT suppliers, notified bodies and regulators.

This paper focuses on the practical aspects of implementation and highlights some of the major changes. It discusses decisions that need to be made by affected organizations and includes questions to ask about your organization’s preparedness in order to comply with the new requirements.

This paper highlights the main areas where ISO 9001:2015 and 13485 have been updated and where they differ; providing Quality Management professionals with the information they need to prepare and plan for the changes in advance...

In order to comply with the European Union (EU) Medical Device Directives – 90/385/EEC Active Implantable MedicalDirectives (AIMD), 93/42/EEC Medical Device Directive (MDD) and 98/79/EC In Vitro Diagnostics Device Directive(IVDD) (referred to as ‘The directives’ hereafter), manufacturers must conduct post-market surveillance (PMS).
